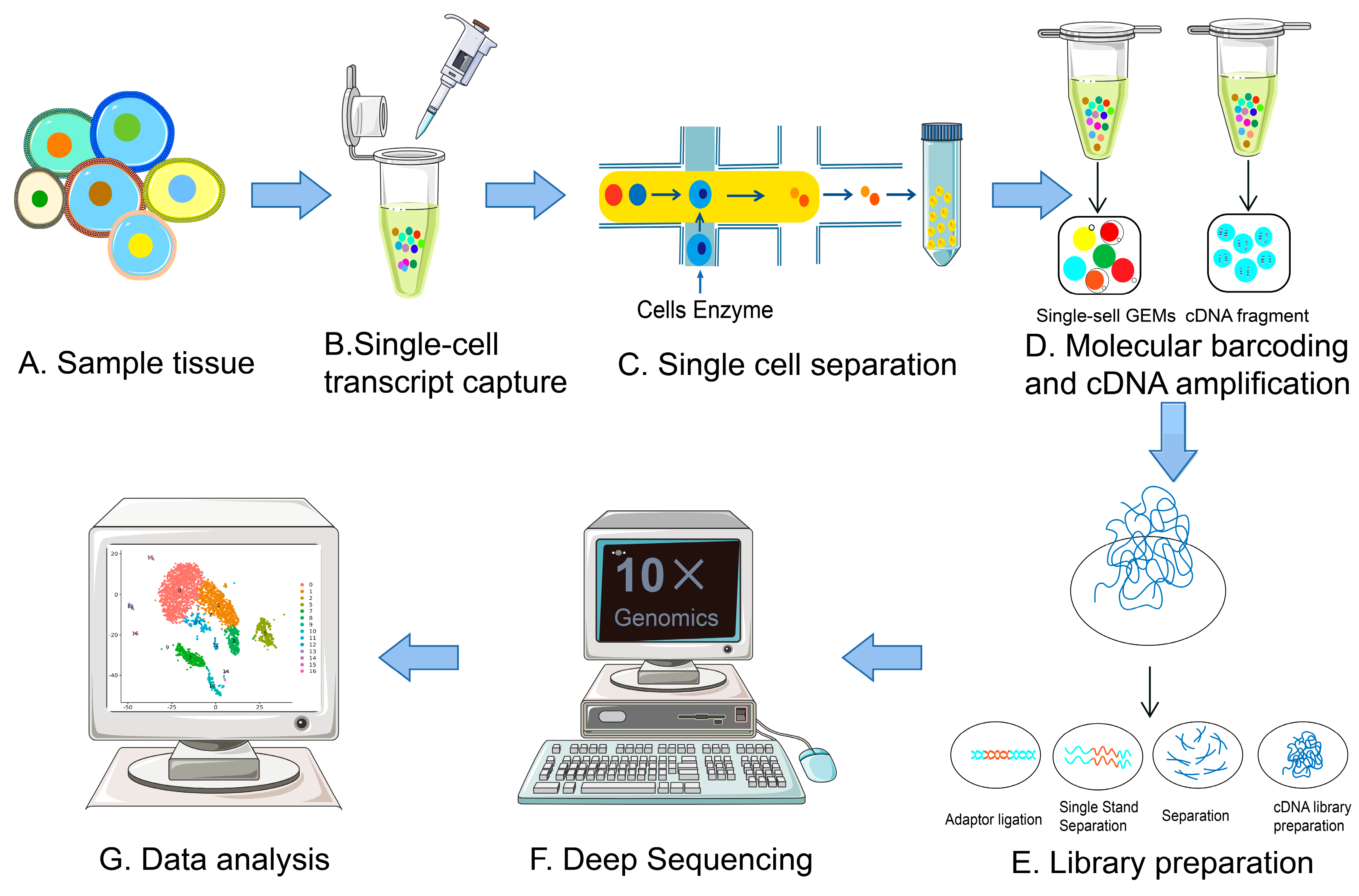
先使用rna-seq的环境
创建metagenomic分析环境
conda create -n metagenomic conda activate metagenomic
安装kneaddata
conda install -c biobakery kneaddata
下载数据 参考: https://blog.csdn.net/Mr_pork/article/details/139743229
这是一个 人类的 结直肠癌 的宏基因组数据,我们选择其中的10个样进行分析
需要数据的文件名,使用prefetch 下载数据,该软件在rna-seq的流程中有
SRA.txt
选择样本的metadata
nohup prefetch -f no --option-file SRA.txt &
可以加 -O 选择输出路径
-O|--output-directory <目录>:保存文件的目录。
直接在超算中输入命令下载,并没有使用sbatch提交作业命令
查看后台任务 SRA文件转为FASTQ格式 单个转格式 慢的转换,太慢了,不建议使用
#将当前 fastq-dump --split-3 --gzip ./SRR12207279
使用这个转换,多线程转换格式,输出的为fq的文件
fasterq-dump --split-3 ./SRR12207283
批量转格式 小命令:删除当前目录SRR文件夹里的所有分文件夹,只保留其文件
find ./SRR -mindepth 1 -type d -exec sh -c 'mv {}/* ./SRR; rmdir {}' \;
fasterq-dump进行批量转换,将所有 .sra 文件都放在SRR文件夹里
sra_dir= "./SRR" output_dir= "./fastq-result" for sra_file in $ sra_dir/ * .srado filename= $ ( basename "$sra_file" .sra) fasterq- dump - - outdir "$output_dir" - - split- 3 "$sra_file" done
生成在当前路径 ./fastq-result 下的 fastq 文件
质控 也只是类似的序列文件,质控流程和转录组相似
fastp质控 此处为扩展学习
conda下载fastp
# note: the fastp version in bioconda may be not the latest conda install -c bioconda fastp
单个
输入 -i -I 双端测序文件 ,输出 -o -O 质控处理后文件,和 json文件,fastp.html结果
fastp -i in.R1.fq.gz -I in.R2.fq.gz -o out.R1.fq.gz -O out.R2.fq.gz
批量
# 创建清理后的文件夹 mkdir clean-fastp # 设置工作目录为fastq文件所在的目录 cd ./fastq-result/ # 遍历所有以_1.fastq结尾的文件 for file1 in *_1.fastq; do # 从文件名中提取没有_1的部分 base=$(basename "$file1" _1.fastq) # 构建对应的_2.fastq文件名 file2="${base}_2.fastq" fileoo1="${base}_1.fq" fileoo2="${base}_2.fq" jsono="${base}.json" htmlo="${base}.html" # 在后台执行检查和trim_galore命令 ( # 检查对应的_2.fastq文件是否存在 if [ -e "$file2" ]; then # 如果存在,执行trim_galore命令 fastp -i "$file1" -o ../clean-fastp/"$fileoo1" -I "$file2" -O ../clean-fastp/"$fileoo2" --json ../clean-fastp/"$jsono" --html ../clean-fastp/"$htmlo" else # 如果不存在,打印错误信息 echo "Error: No matching file found for $file1" fi ) & done # 等待所有后台进程完成 wait
multiqc汇总质控结果 multiqc ./fastq-result/ -o ./fastq-result/
这个结果看不看无所谓,fastp是强大的质控软件,只要输入文件无误,结果也不会有问题
.fq 结尾的文件即为之后进行分析的,清洗之后的序列文件
去宿主-方法一 去宿主的过程其实就是将序列比对到宿主基因组上,然后没有比对到的序列整合成新文件就是去宿主后的了。宿主基因组需要自己先下载好并用 bowtie2-build 建立索引,以人类为例:
构建索引 在官网中找到自己的物种 https://hgdownload.soe.ucsc.edu/downloads.html
人的基因组 hg38
http://hgdownload.cse.ucsc.edu/goldenPath/hg38/bigZips/hg38.chromFa.tar.gz
鼠的基因组 mm10
http://hgdownload.cse.ucsc.edu/goldenPath/mm10/bigZips/chromFa.tar.gz
wget http://hgdownload.cse.ucsc.edu/goldenPath/hg38/bigZips/hg38.chromFa.tar.gz tar -zxvf chromFa.tar.gz cat *.fa > hg38.fa bowtie2-build hg38.fa hg38
自己构建索引有点慢
bowtie2比对,单个 参考 https://www.jianshu.com/p/fe9c5cc7373e
bowtie2 -p 20 -x /public/home/dk_szy/songxudong/metagenomic/db/hg37dec_v0.1 -1 data/fastp/${sample}_1.fq.gz \ -2 data/fastp/${sample}_2.fq.gz -S data/rm_human/${sample}.sam \ --un-conc data/rm_human/${sample}.fq --very-sensitive rm data/rm_human/${sample}.sam
bowtie2比对,批量 # 将工作目录设置为fastq文件所在的目录!!!! mkdir rm_human cd ./clean-fastp/ # 将传入的参数赋值给变量!!!!! file1_pattern="_1.fq" file2_pattern="_2.fq" # 遍历所有以第一个参数模式结尾的文件 for file1 in *${file1_pattern}; do # 从文件名中提取去掉模式后的部分 base=$(basename "$file1" ${file1_pattern}) # 构建对应的第二个参数模式的文件名 file2="${base}${file2_pattern}" # 在后台执行检查和trim_galore命令 ( # 检查对应的文件是否存在 if [ -e "$file2" ]; then echo "找到名为 "$base" 的文件 $file1 对应 $file2 " # 如果存在,例如,执行trim_galore命令!!!!!! bowtie2 -p 20 -x /public/home/dk_szy/songxudong/metagenomic/db/hg37dec_v0.1 -1 "$file1" \ -2 "$file2" -S ${base}.sam \ --un-conc ../rm_human/${base}.fq --very-sensitive rm ${base}.sam else # 如果不存在,打印错误信息 echo "错误: 未找到与 $file1 匹配的文件" fi ) & done # 等待所有后台进程完成 wait
#使用hisat2直接用构建好的索引进行比对 尝试使用建立好的索引进行比对
mkdir -p ./align/flag cd ./align/ pwd ##参考基因组的位置 index='/public/home/dk_szy/songxudong/rna-test/reference/human-UCSC-hg38/hg38/genome' # 假设你的fastq文件在fastq-result文件夹中 fastq_dir="../clean-fastp" # 遍历fastq-result文件夹中的所有1.fastq文件 for file1 in $fastq_dir/*_1.fq; do # 从1.fastq文件名中提取ID,并删除_1_val_1 id=$(basename "$file1" .fq | sed 's/_1_val_1//') # 查找对应的2.fastq文件 file2="$fastq_dir/${id}_2.fq" # 检查2.fastq文件是否存在 if [ -f "$file2" ]; then echo "333# ${id} !!!!! is on the hisat2 Working !!!" # 使用hisat2进行比对,并指定输出目录为当前目录(./align/) hisat2 -t -p 20 -x $index \ -1 "$file1" \ -2 "$file2" -S "${id}.sam" # sam2bam and remove sam,指定输出目录为当前目录(./align/) echo -e " ${id} sam2bam and remove sam " samtools view -F 12 -@ 12 -b "./${id}.sam" > "./${id}_sorted.bam" rm "./${id}.sam" else echo "No matching 2.fastq file found for $file1" fi done
#去宿主-方法二 kneaddata 使用 kneaddata进行质控和去宿主
kneaddata自带的参考序列有限,主要为人和小鼠,其他的需要自己构建
#查看有哪些数据库 kneaddata_database
下载人的参考序列,速度似乎还可以
mkdir -p db kneaddata_database --download human_genome bowtie2 db/
kneaddata -i seq/C2_1.fq.gz -i seq/C2_2.fq.gz -o qc/ -v -t 8 --remove-intermediate-output --trimmomatic ~/.conda/envs/qc2/share/trimmomatic --trimmomatic-options 'ILLUMINACLIP:~/.conda/envs/qc2/share/trimmomatic/adapters/TruSeq3-PE.fa:2:40:15 SLIDINGWINDOW:4:20 MINLEN:50' --bowtie2-options '--very-sensitive --dovetail' --bowtie2-options="--reorder" -db db/Homo_sapiens
物种注释:kraken2 参考:https://www.jianshu.com/p/fe9c5cc7373e
Kraken2是一个用于对高通量测序数据进行分类和标识物种的软件。它使用参考数据库中的基因组序列来进行分类,并使用k-mer方法来实现快速和准确的分类。
使用Kraken2进行基本分类的简单步骤:
安装Kraken2:可以从Kraken2官方网站下载并安装Kraken2软件。
conda install bioconda::kraken2
准备参考数据库:Kraken2需要一个参考数据库,以便对测序数据进行分类。可以直接下载官方构建的标准库,也可以从NCBI、Ensembl或其他数据库下载相应的基因组序列,并使用Kraken2内置的工具来构建数据库。
--standard标准模式下只下载5种数据库:古菌archaea、细菌bacteria、人类human、载体UniVec_Core、病毒viral。
#超算中未成功,选择自行下载 #kraken2-build --standard --threads 20 --db ./
选择自行下载网站: https://benlangmead.github.io/aws-indexes/k2
下载Standard 文件大小 90G
#自行下载命令 wget -c https://genome-idx.s3.amazonaws.com/kraken/k2_standard_20240605.tar.gz
#运行Kraken2:使用Kraken2对测序数据进行分类需要使用以下命令:
kraken2 --db <path_to_database> <input_file> --output <output_file>
这里,<path_to_database>是参考数据库的路径,<input_file>是需要进行分类的输入文件,<output_file>是输出文件的名称。Kraken2将输出一个分类报告文件和一个序列文件。
需要注意的是kraken运行至少要提供数据库大小的内存大小(运行内存),因为它会把整个数据库载入内存后进行序列的注释,所以如果发现无法载入数据库的报错,可以尝试调大内存资源。
单个比对 第二行- -db 为存放参考索引的文件夹
kraken2 --threads 20 \ --db /public/home/dk_szy/songxudong/metagenomic/test \ --confidence 0.05 \ --output ./result/test.output \ --report ./report/test.kreport \ --paired \ ../rm_human/ERR1018185.1.fq \ ../rm_human/ERR1018185.2.fq
批量比对 mkdir kraken-result # 将工作目录设置为fastq文件所在的目录!!!! cd ./rm_human/ # 将传入的参数赋值给变量!!!!! file1_pattern=".1.fq" file2_pattern=".2.fq" # 遍历所有以第一个参数模式结尾的文件 for file1 in *${file1_pattern}; do # 从文件名中提取去掉模式后的部分 base=$(basename "$file1" ${file1_pattern}) # 构建对应的第二个参数模式的文件名 file2="${base}${file2_pattern}" # 在后台执行检查和trim_galore命令 ( # 检查对应的文件是否存在 if [ -e "$file2" ]; then echo "找到名为 "$base" 的文件 $file1 对应 $file2 " # 如果存在,例如,执行trim_galore命令!!!!!! #trim_galore -q 25 --phred33 --length 35 --stringency 3 --paired -o ../clean_data/ "$file1" "$file2" kraken2 --threads 20 \ --db /public/home/dk_szy/songxudong/metagenomic/test \ --confidence 0.05 \ --output ../kraken-result/${base}.output \ --report ../kraken-result/${base}.kreport \ --paired \ ./${file1} \ ./${file2} echo "文件 "$base" 运行完毕 " else # 如果不存在,打印错误信息 echo "错误: 未找到与 $file1 匹配的文件" fi ) & done # 等待所有后台进程完成 wait
物种组成及丰度估计 只能说宏基因组的教程也太水了,未找到对结果的处理部分
使用教程:https://www.jianshu.com/nb/54122549
运行 bracken 进行各个分类水平物种丰度估计:
先安装 bracken
conda install bioconda::bracken
运行 bracken 进行各个分类水平物种丰度估计:
mkdir out # 将工作目录设置为fastq文件所在的目录 cd ./kraken-result/ # 将传入的参数赋值给变量 file1_pattern=".kreport" # 遍历所有以第一个参数模式结尾的文件 for file1 in *${file1_pattern}; do # 从文件名中提取去掉模式后的部分 base=$(basename "$file1" ${file1_pattern}) # 直接执行trim_galore命令,不需要检查对应的file2是否存在 ( echo "找到名为 $base 的文件 $file1" #循环执行代码区 # 运行bracken bracken \ -d /public/home/dk_szy/songxudong/metagenomic/test \ -i ${base}.kreport \ -o ../out/${base}.bracken.S \ -w ../out/${base}.bracken.S.kreport \ -l S \ -t 20 ) & done # 等待所有后台进程完成 wait
结果整理 安装kraken-biom
conda install bioconda::kraken-biom
# report文件合并成biom格式 kraken-biom \ ./out/*.kreport \ --max D \ -o ./out/S.biom # biom转count表格 # 注意:这里假设convert是指向biom转换为tsv的工具,不是ImageMagick的convert # 如果是ImageMagick的convert,那么下面的命令是错误的 # 正确的命令应该是直接使用biom工具进行转换 biom convert \ -i ./out/S.biom \ -o ./out/S.count.tsv.tmp \ --to-tsv \ --header-key taxonomy # 输出文件格式调整,补全物种名 sed 's/; g__; s__/; g__; s__ /' ./out/S.count.tsv.tmp \ > ./out/S.taxID.count.tsv # taxonID 替换回拉丁名 sed '/^#/! s/^[[0-9]]+\t\(.*[A-Za-z]__\([^\t;]\+\)\)$/\2\t\1/' \ ./out/S.taxID.count.tsv > ./out/S.taxName.count.tsv # 保留丰度信息,用于后续绘图 sed '1d; 2s/^#//' ./out/S.taxName.count.tsv | \ awk -F $'\t' -v 'OFS=\t' '{$NF = ""; print $0}' | \ sed 's/\t$//' > ./out/S.count.tsv
输出结果:
暂未准备绘图代码
a多样性 教程中未具体讲解方法,但是提到 使用 vegan 及 phyloseq 两个 R 包进行多样性分析
使用16s做过的方法进行分析
OTU文件 S.count.tsv
分组文件 group.txt
R语言
getwd( ) setwd( "D:/rtest/songtest/metagenomic" ) if ( ! requireNamespace( "BiocManager" , quietly = TRUE ) ) install.packages( "BiocManager" ) BiocManager:: install( "phyloseq" ) rm( list = ls( ) ) library( vegan) library( reshape2) library( ggplot2) library( ggpubr) library( RColorBrewer) data <- read.delim( "D:/rtest/songtest/metagenomic/S.count.txt" , header= TRUE , sep= "\t" , row.names= 1 ) group <- read.delim( "D:/rtest/songtest/metagenomic/group.txt" ) otu <- data colSums( data) otu_Flattening = as.data.frame( t( rrarefy( t( otu) , min ( colSums( otu) ) ) ) ) colSums( otu_Flattening) data <- otu_Flattening ttdata <- t( data) data<- data/ apply( data, 2 , sum ) tdata= t( data) a<- as.data.frame( tdata) a= as.data.frame( lapply( a, as.numeric ) ) shannon<- diversity( a, index= "shannon" ) simpson<- diversity( a, index= "simpson" ) Chao1 <- estimateR( ttdata) [ 2 , ] ACE <- estimateR( ttdata) [ 4 , ] invsimpson<- diversity( a, index= "invsimpson" ) data_shannon= data.frame( shannon) data_simpson= data.frame( simpson) data_Chao1= data.frame( Chao1) data_ACE= data.frame( ACE) data_invsimpson= data.frame( invsimpson) spe_alpha<- cbind( data_shannon, data_simpson, data_invsimpson, data_Chao1, data_ACE, group )
物种分类结果整理 还是使用之前16s的方法
使用上面的 S.taxName.count.tsv 为包含物种分类信息的丰度文件,可以用此结果绘制物种堆积图
setwd( "D:/rtest/songtest/metagenomic/lefse" ) taxon_data <- read.delim( "D:/rtest/songtest/metagenomic/lefse/S.taxName.count.tsv" , row.names= 1 ) taxon_data$ kingdom <- sapply( strsplit( taxon_data$ taxonomy, ";" ) , `[`, 1 ) taxon_data$ phylum <- sapply( strsplit( taxon_data$ taxonomy, ";" ) , `[`, 2 ) taxon_data$ class <- sapply( strsplit( taxon_data$ taxonomy, ";" ) , `[`, 3 ) taxon_data$ order <- sapply( strsplit( taxon_data$ taxonomy, ";" ) , `[`, 4 ) taxon_data$ family <- sapply( strsplit( taxon_data$ taxonomy, ";" ) , `[`, 5 ) taxon_data$ genus <- sapply( strsplit( taxon_data$ taxonomy, ";" ) , `[`, 6 ) taxon_data$ species <- sapply( strsplit( taxon_data$ taxonomy, ";" ) , `[`, 7 ) write.csv( taxon_data, file = "otu-result.csv" )
导出分类好的 otu-result.csv 文件
得到每个水平的结果 界(Kingdom)、门(Phylum)、纲(Class)、目(Order)、科(Family)、属(Genus)、种(Species)
16s只能注释到 属(Genus)水平的结果,虽然我们有属水平的文件,但是是没有相应的可用结果
library( dplyr) otu_data <- read.csv( "otu-result.csv" , header = TRUE , row.names = 1 ) otu_data <- otu_data[ ! names ( otu_data) %in% "Confidence" ] otu_data <- otu_data[ otu_data$ kingdom != "Unassigned" , ] taxonomic_levels <- c ( "phylum" , "class" , "order" , "family" , "genus" , "species" ) previous_level <- "kingdom" for ( i in seq_along ( taxonomic_levels) ) { current_level <- taxonomic_levels[ i] otu_data[[ current_level] ] <- as.character ( otu_data[[ current_level] ] ) otu_data[[ previous_level] ] <- as.character ( otu_data[[ previous_level] ] ) summary_data <- otu_data %>% group_by( ! ! sym( previous_level) , ! ! sym( current_level) ) %>% summarise( across( where( is.numeric ) , sum , na.rm = TRUE ) ) %>% ungroup( ) csv_filename <- paste0( previous_level, "_" , current_level, ".csv" ) write.csv( summary_data, csv_filename, row.names = FALSE ) previous_level <- current_level }
基因组组装 https://www.jianshu.com/p/77131fa96caa
使用 megahit 进行组装:
安装
conda activate metagenomic conda install bioconda::megahit
单个的示例
megahit \ -1 ./A1_1.fq.gz \ # 输入,fq1 -2 ./A1_2.fq.gz \ # 输入,fq2 --min-contig-len 1000 \ # contig最小长度 --tmp-dir ./ \ # 设置tmp目录 --memory 6 \ # 内存占用 --num-cpu-threads 4 \ # 线程数 --out -dir A1_megahit \ # 输出目录 --out -prefix A1 # 输出前缀 ## 多组数据组装, 输入数据逗号分隔
批量组装 要求:输出文件夹会自动创建,开始时不需要存在
# 将工作目录设置为fastq文件所在的目录!!!! cd ./fastq-result/ # 将传入的参数赋值给变量!!!!! file1_pattern=".1.fq" file2_pattern=".2.fq" #${file1} #${file2} #${base} # 遍历所有以第一个参数模式结尾的文件 for file1 in *${file1_pattern}; do # 从文件名中提取去掉模式后的部分 base=$(basename "$file1" ${file1_pattern}) # 构建对应的第二个参数模式的文件名 file2="${base}${file2_pattern}" # 在后台执行检查和trim_galore命令 ( # 检查对应的文件是否存在 if [ -e "$file2" ]; then echo "找到名为 "$base" 的文件 $file1 对应 $file2 " # 如果存在,例如,执行trim_galore命令!!!!!! #trim_galore -q 25 --phred33 --length 35 --stringency 3 --paired -o ../clean_data/ "$file1" "$file2" megahit \ -1 ./rm_human/${file1} \ -2 ./rm_human/${file2} \ --min-contig-len 1000 \ --tmp-dir ./ \ --memory 6 \ --num-cpu-threads 20 \ --out-dir ./megahit-result/ \ --out-prefix ${base} else # 如果不存在,打印错误信息 echo "错误: 未找到与 $file1 匹配的文件" fi ) & done # 等待所有后台进程完成 wait
Lefse 参考: https://blog.csdn.net/a852232394/article/details/139296579?spm=1001.2101.3001.6650.2&utm_medium=distribute.pc_relevant.none-task-blog-2%7Edefault%7EYuanLiJiHua%7EPosition-2-139296579-blog-126683847.235%5Ev43%5Econtrol&depth_1-utm_source=distribute.pc_relevant.none-task-blog-2%7Edefault%7EYuanLiJiHua%7EPosition-2-139296579-blog-126683847.235%5Ev43%5Econtrol&utm_relevant_index=5
需要3个输入文件
sample_table.csv
feature_table.csv
tax_table.csv
rm(list=ls()) pacman::p_load(tidyverse,microeco,magrittr) feature_table <- read.csv('feature_table.csv', row.names = 1) sample_table <- read.csv('sample_table.csv', row.names = 1) tax_table <- read.csv('tax_table.csv', row.names = 1) head(feature_table)[,1:6]; head(sample_table); head(tax_table)[,1:6] dataset <- microtable$new(sample_table = sample_table, otu_table = feature_table, tax_table = tax_table) dataset lefse <- trans_diff$new(dataset = dataset, method = "lefse", group = "Group", #过少可增大下面选项 alpha = 0.1, lefse_subgroup = NULL) write.csv(lefse$res_diff,file = "lefse-lda.csv") #保存 write.csv(lefse$abund_table,file = "lefse-input.csv") #保存
分析出初步结果
lefse-lda.csv LDA,可自行画图
lefse-input.csv lefse输入文件,可在在线网站进行可视化或者R可视化
在线绘制 复制 lefse-input.csv 内容,进行修改
https://www.bic.ac.cn/BIC/#/
找到lefse选项
主要是更改第一行的列名,定义分组
检查数据可用后可直接出结果
(某些未知的菌可在原始数据中提前删除)
在线网站的编辑功能好像更强大了,作图方便建议采用
R绘制 https://blog.csdn.net/a852232394/article/details/139296579?spm=1001.2101.3001.6650.2&utm_medium=distribute.pc_relevant.none-task-blog-2~default~YuanLiJiHua~Position-2-139296579-blog-126683847.235%5Ev43%5Econtrol&depth_1-utm_source=distribute.pc_relevant.none-task-blog-2~default~YuanLiJiHua~Position-2-139296579-blog-126683847.235%5Ev43%5Econtrol&utm_relevant_index=5
使用 microeco包 自带的绘图,并不算好看,可进行美化或使用原始数据自行绘图
LDA
##use_number 为显示的个数 ##group_order 为自己的分组 lefse$plot_diff_bar(use_number = 1:20, width = 0.8, group_order = c("subject-1", "subject-2"))
lefse
library(ggtree) lefse$plot_diff_cladogram(use_taxa_num = 200, use_feature_num = 50, clade_label_level = 5, group_order = c("subject-1", "subject-2"))