AutoDock分子对接
AutoDock分子对接
Song Xudong文件下载
下载PDB文件
以eEF2K和橄榄苦苷为例进行对接
eEF2K的文件,eEF2K-TR,无ATP,ADP的结构,包含CaM
https://www.rcsb.org/structure/7SHQ
橄榄苦苷的文件Oleuropein
https://pubchem.ncbi.nlm.nih.gov/compound/5281544
将文件放在Autodock工作目录(不能有中文路径)
格式转化
使用Open Babel将Oleuropein的sdf文件转化为autodock可用的pdb文件
选择input和output的格式和目录,点击CONVERT进行转换

Autodock对接
软件启动
记得关闭AMD显卡和联想电脑管家
参考:https://www.zhihu.com/question/393216168
对接流程
参考:https://zhuanlan.zhihu.com/p/662465038
打开autodock软件
首先,设置路径,file->perfernce->set,出现下图界面,选择startup directory,把我们刚刚含有5个文件的文件路径拷贝进来。
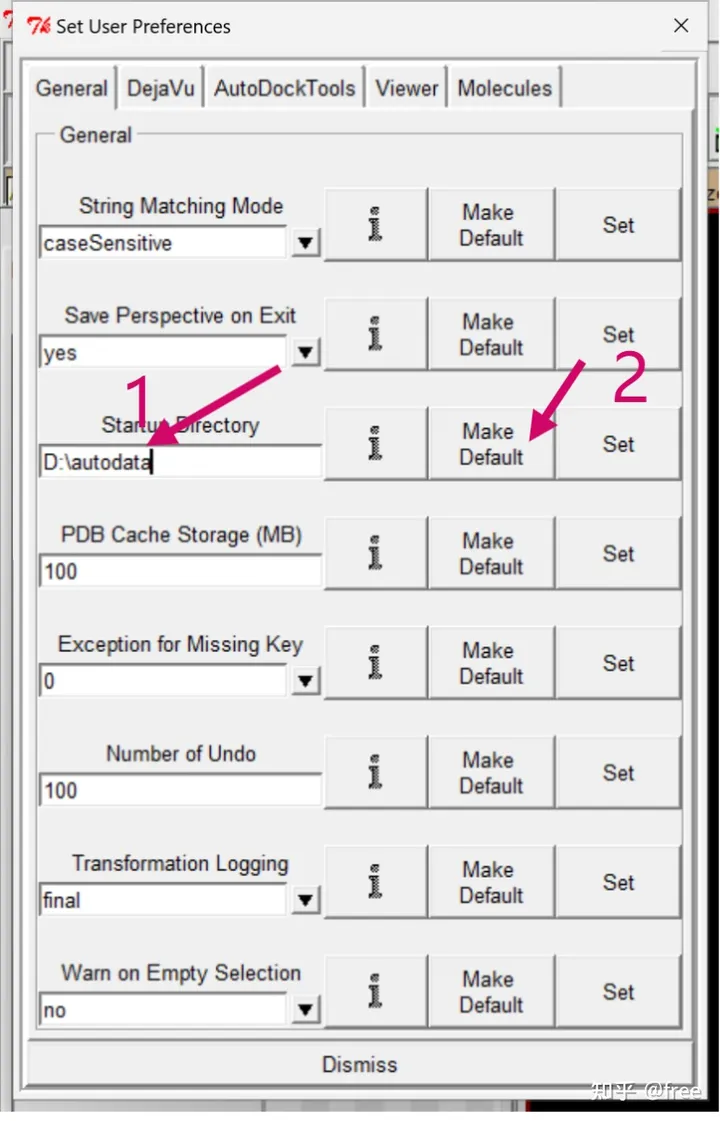
然后点击Make Default,将它设置为默认路径。
3.对蛋白质进行前处理。点击file->read molecule,选择蛋白质分子
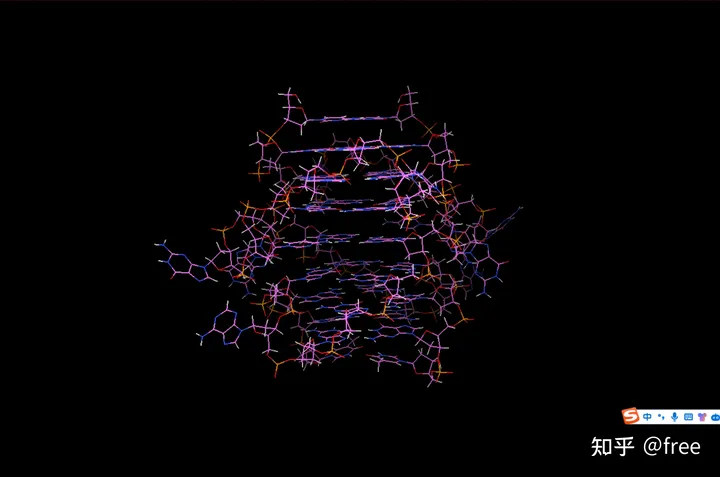
首先,对蛋白质进行除水加氢键。点击edit->delete water,edit->hydrogens->add->ok
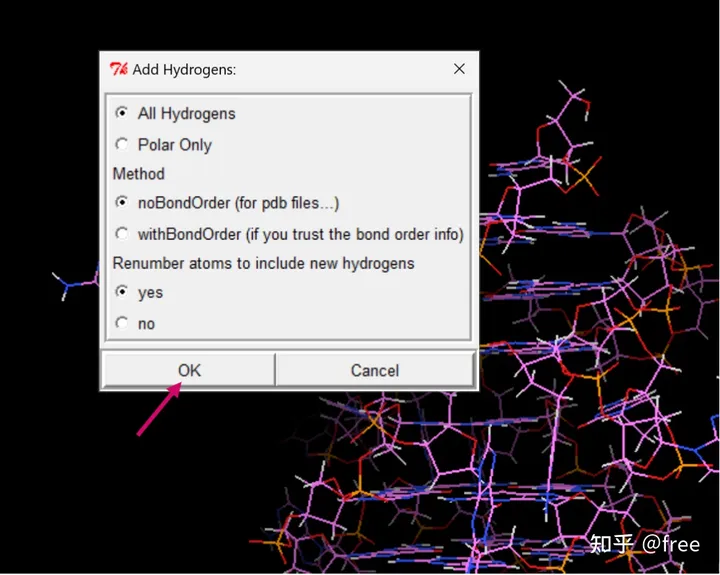
把它选择为大分子,点击grid->macromolecules->choose,选择大分子,点击select molecule
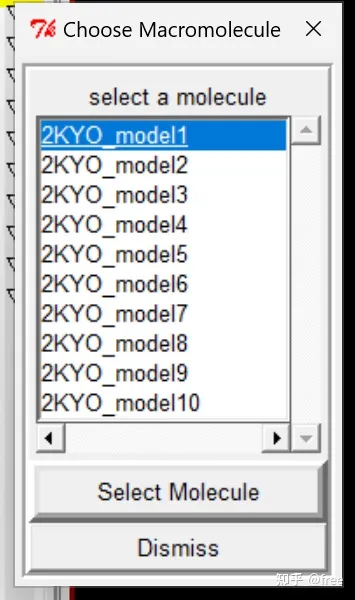
得到蛋白质的pdbqt的一个文件,保存即可(注意不要出现特殊符号,下图中的—就有可能导致运行错误)
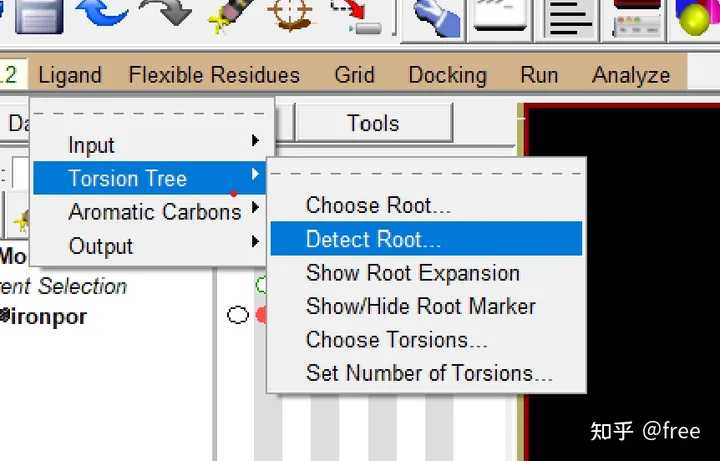
4.对小分子处理,然后把蛋白质删掉,edit->delete,导入小分子,与蛋白质导入相同的步骤,同样的进行加氢处理,ligand->input->choose,选择小分子作为ligand,同时对扭转键进行检测。ligand->torsion tree->detect root
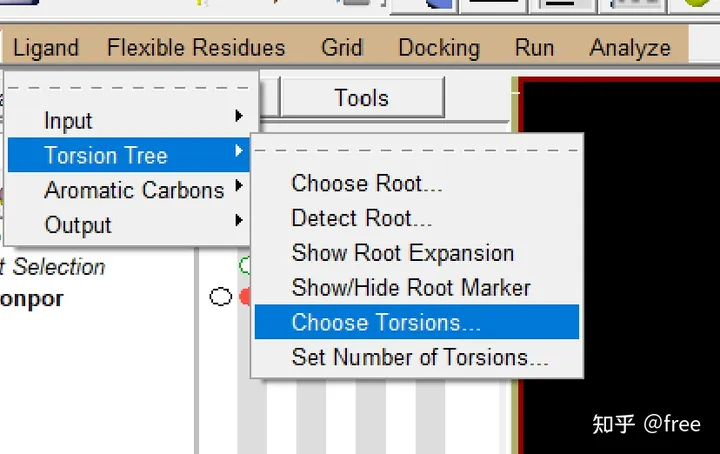
ligand->torsion tree->choose torsions
done
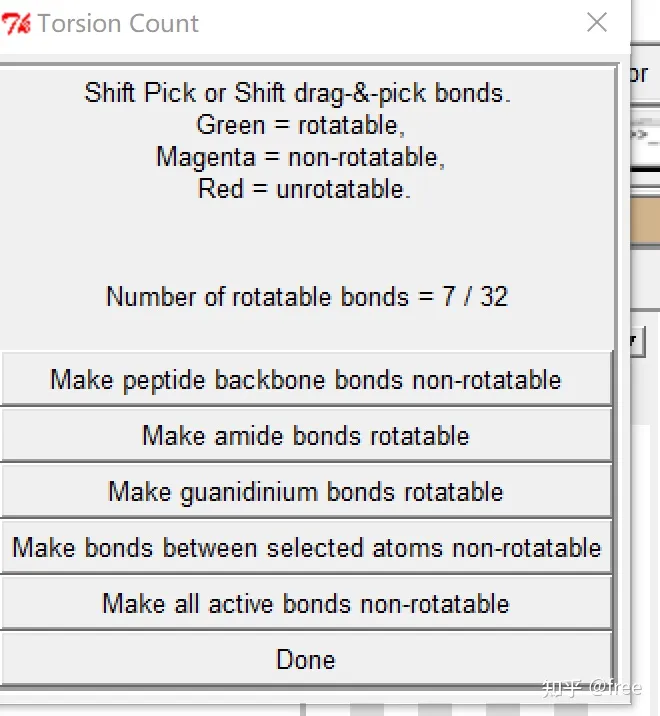
红色的是不可以被扭转的,绿色的是可以扭转的

输出小分子,ligand->output->save as PDBQT,得到小分子的pdbqt格式的文件。
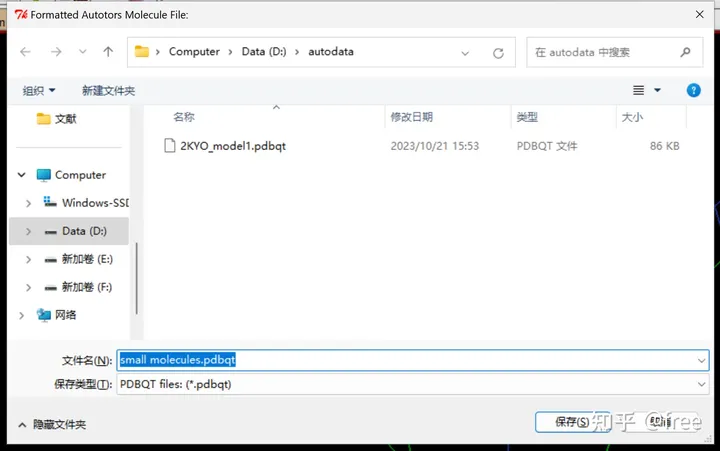
四,开始对接。
删掉当前的小分子。
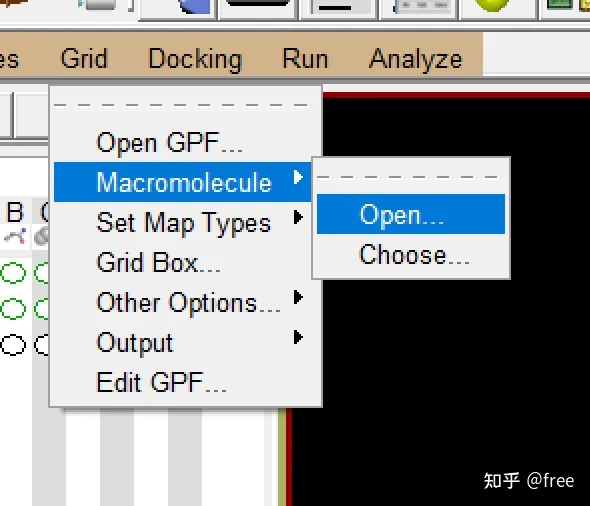
先把蛋白质导入进来,点击Grid->macromolecules->open,选择蛋白质大分子。后面出现的选项全部选择yes,确定。
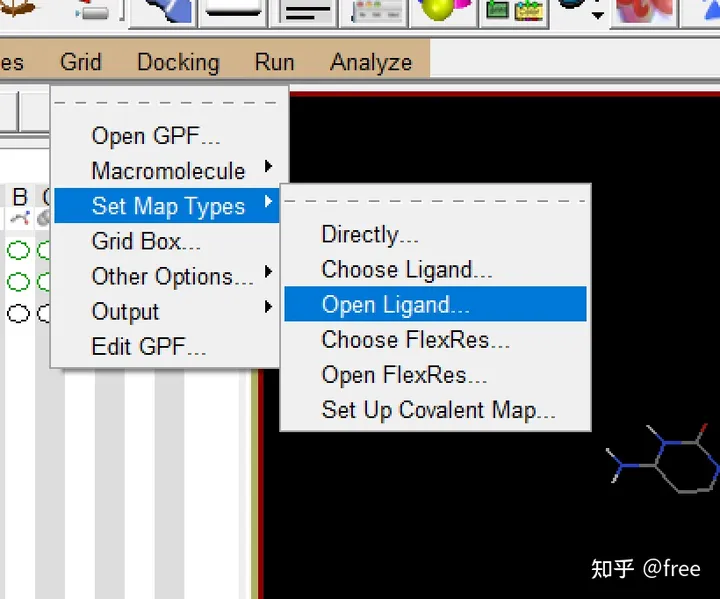
接着导入小分子,Grid->set map types->open ligand
此时开始对接,对参数进行一些设置,点击grid->grid box出现一个立方体
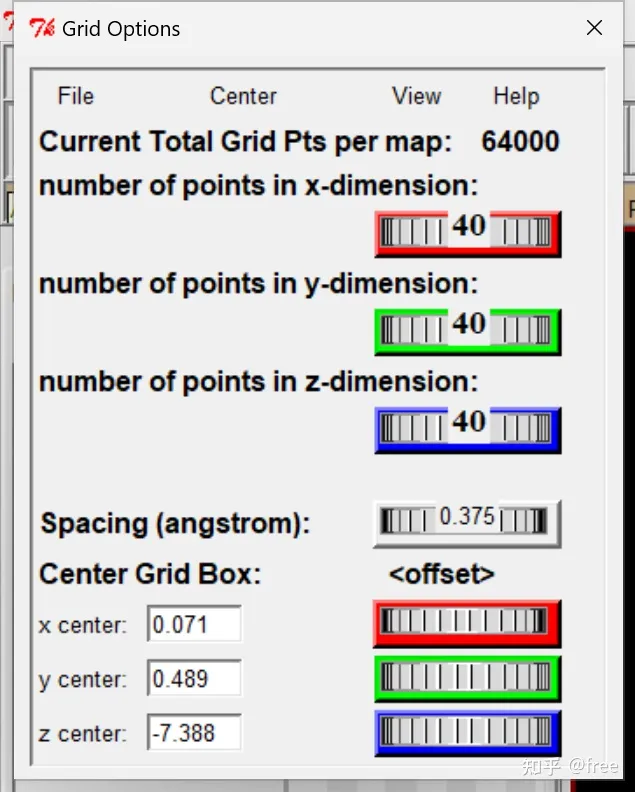
由于我们不知道大概的结合位点在哪里,需要调节这个三位参数,把蛋白质和小分子都包含在内,可以通过旋转观察否包含在内。
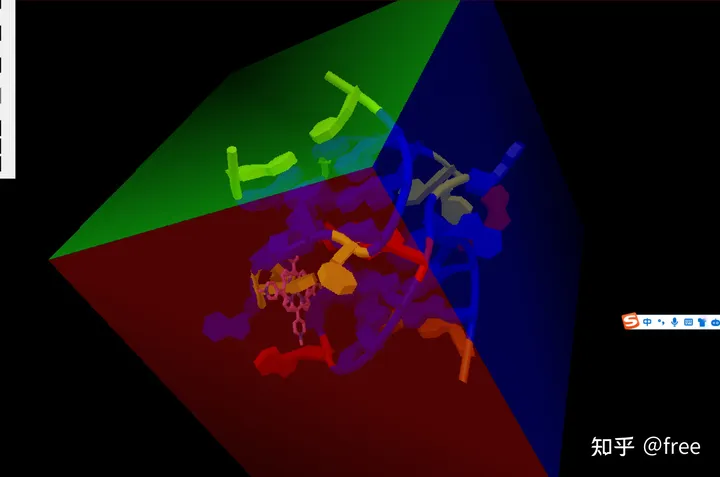
到这个程度就可以了。
点击dejavu gui,即这个图标

点击root,选择小分子,把图中的对钩取消勾选
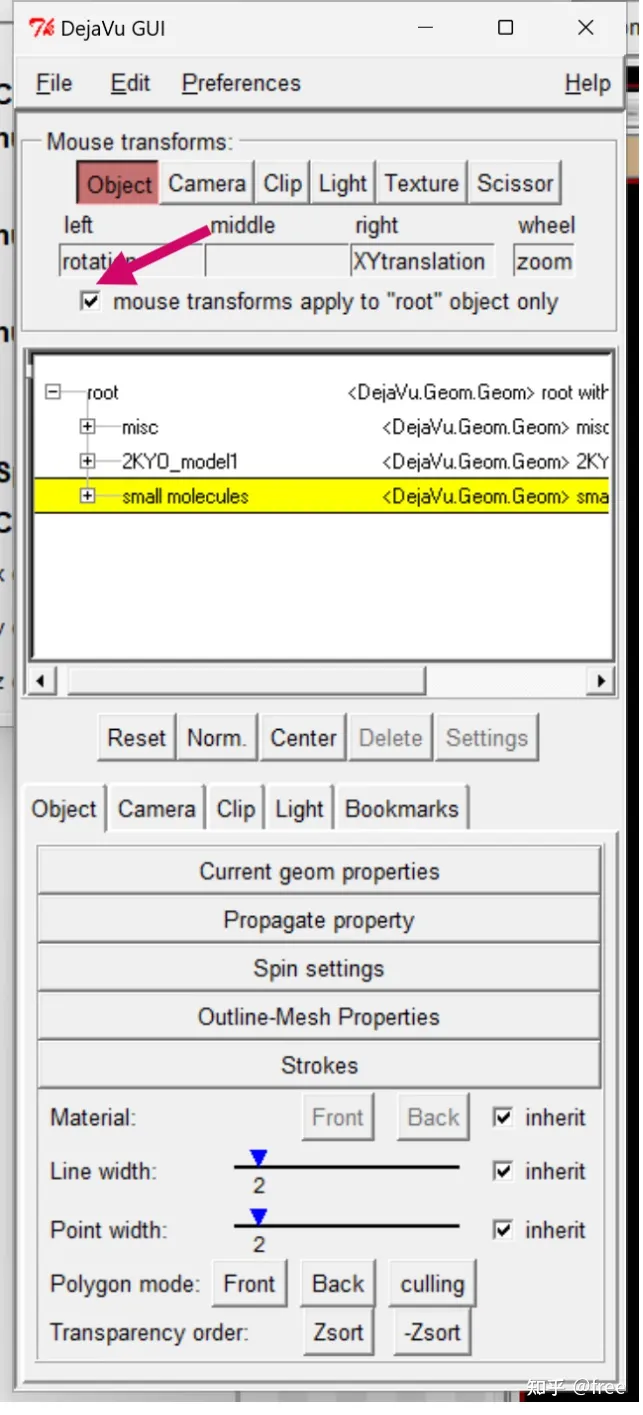
此时,用右键把小分子拖出到立方体外,此时,再把刚刚取消勾选的√给勾上。再点击刚刚打开窗口的file,close saving current
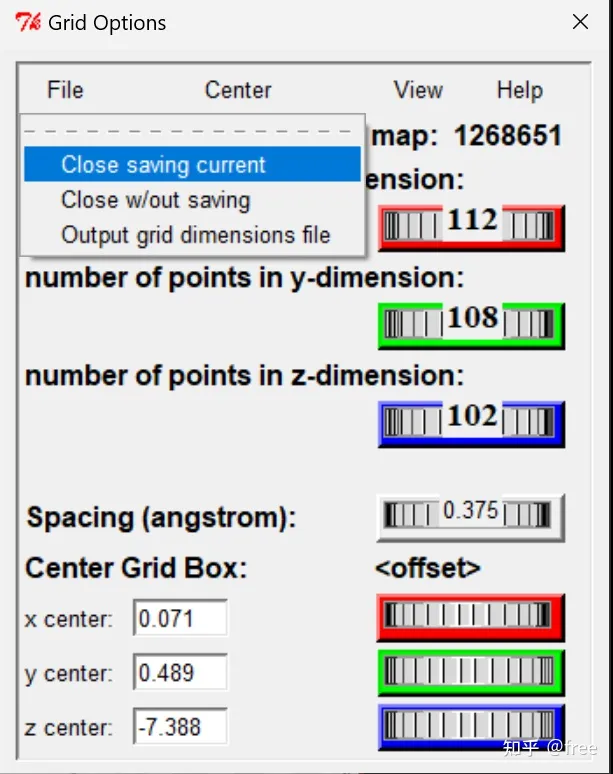
点击grid->output->save GPF
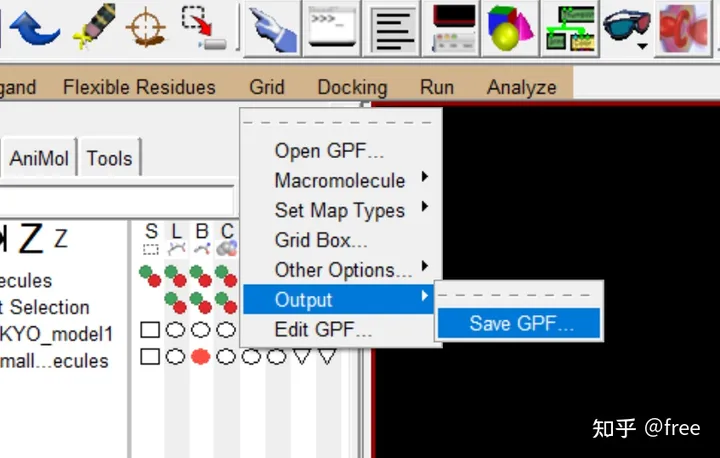
保存为1,后缀如果不能正常出现的话,手动输入后缀gpf。
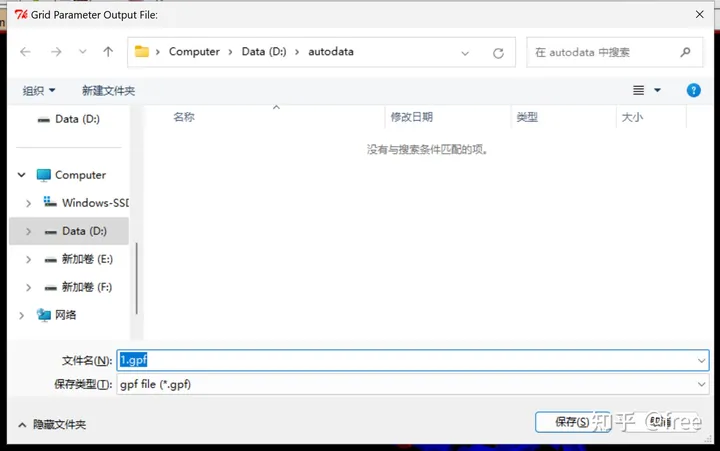
点击保存(注意,不要出现中文字符或者空格以及特殊符号)
点击run->autogrid
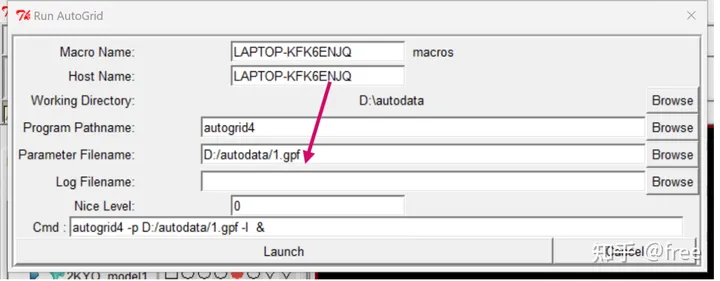
browse 我们的gpf文件
会生成一个glg文件
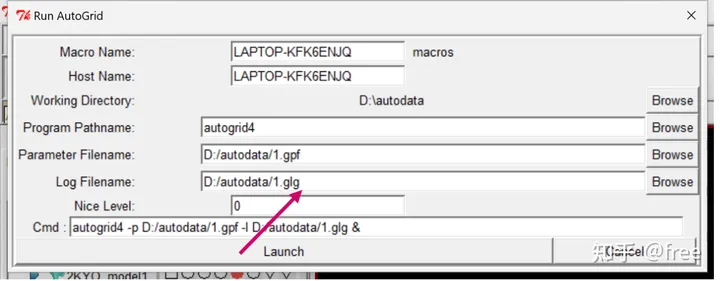
点击launch,生成一个新窗口,等待这个窗口运行完毕。
运行完毕,数据文件夹中会多出很多map结尾的文件,还有一个glg文件。
点击Docking->macromolecules->set rigid file name,打开蛋白质大分子。再点击docking-》ligand->choose->选择小分子->set as ligand,然后点击接受。
点击Docking->search parameters->genetic algorithm
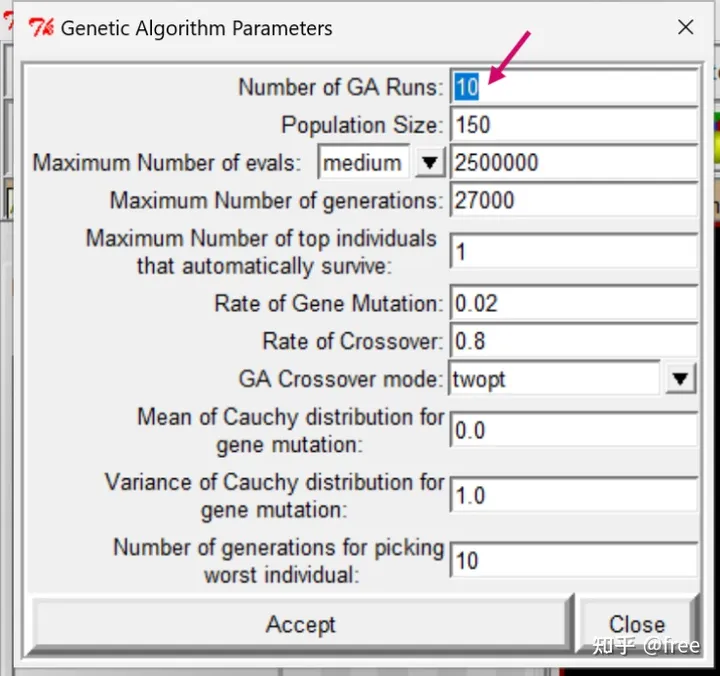
第一排是对接次数,我这里选择50次(官方建议对接50次),点击accept。
接着Docking->docking parameters->accept。
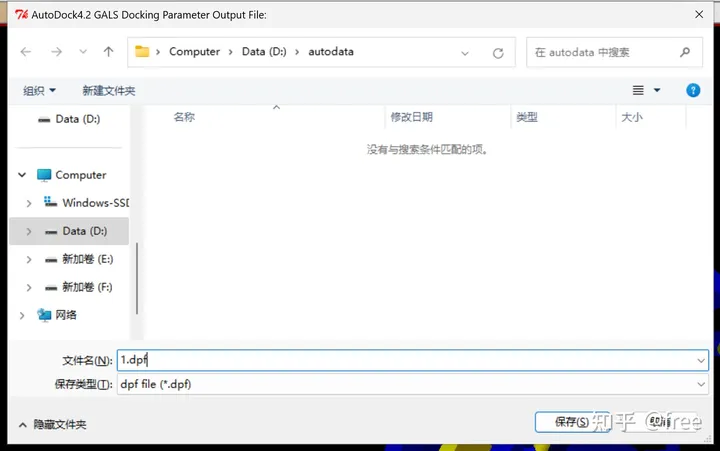
docking->output->lamarckian(4.2),输出文件,后缀同样手动添加dpf,点击保存。
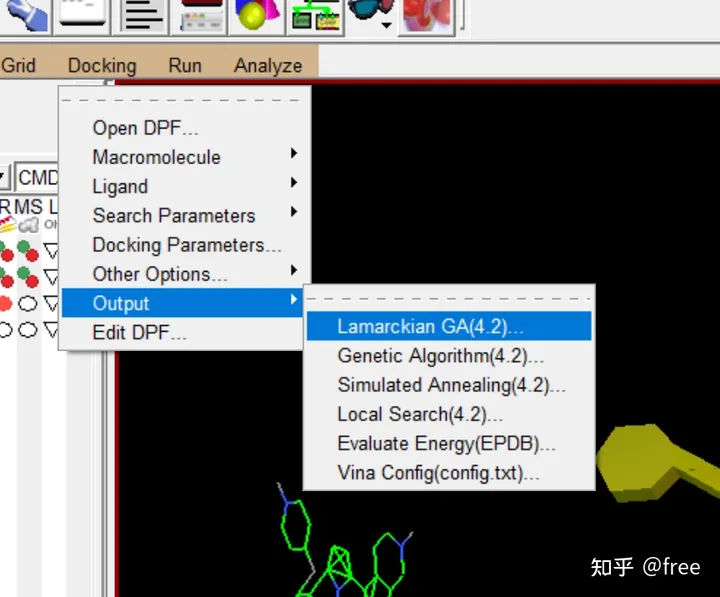
点击run->run autodock
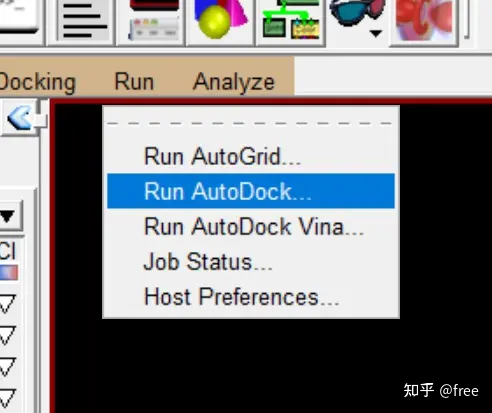
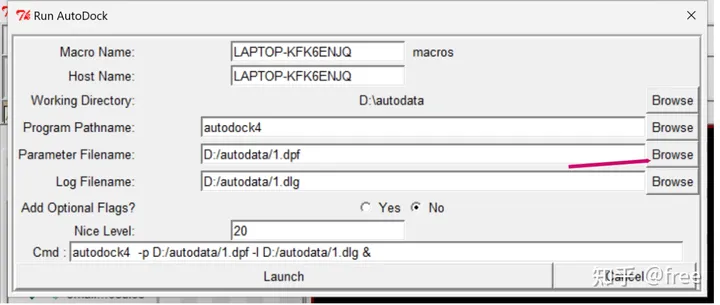
browse刚刚保存的dpf文件,生成一个dlg文件,点击launch,等待程序运行。(对接时间较长,框体自动关闭为完成)
文件夹中dlg文件生成好之后,可以删掉所有的分子,点击edit->delete->all molecules.
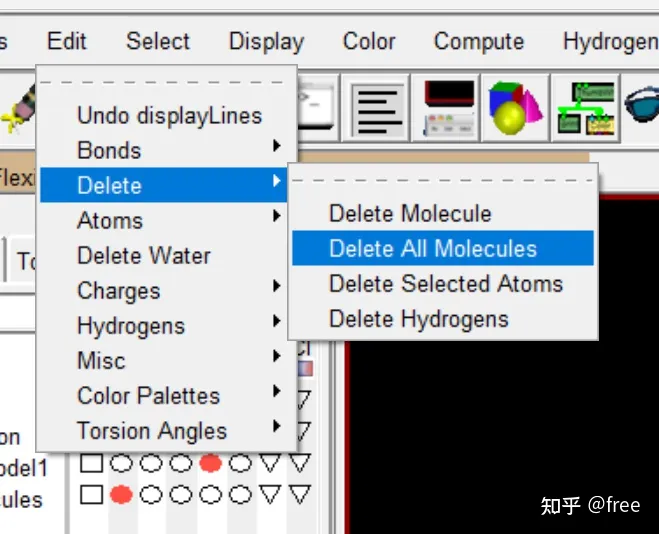
五、对结果进行分析
analysis->docking->open
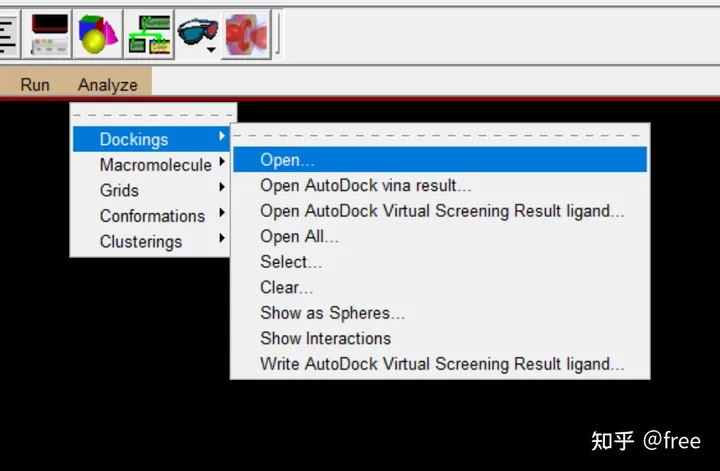
打开dlg文件,点击确定,点击analyze-》macromolecule->open,等待大分子出现,接着点击analyze-》conformations-》play ranked by enanergy,出现一个新窗口如下图
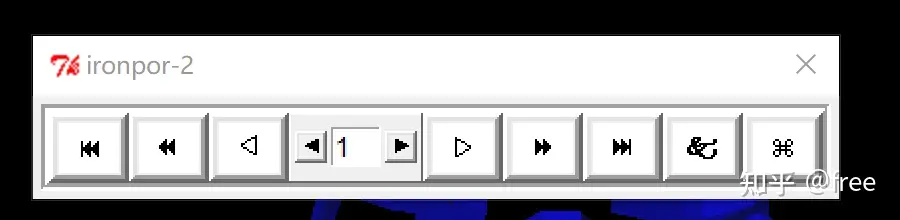
点击倒数第二个按钮,即

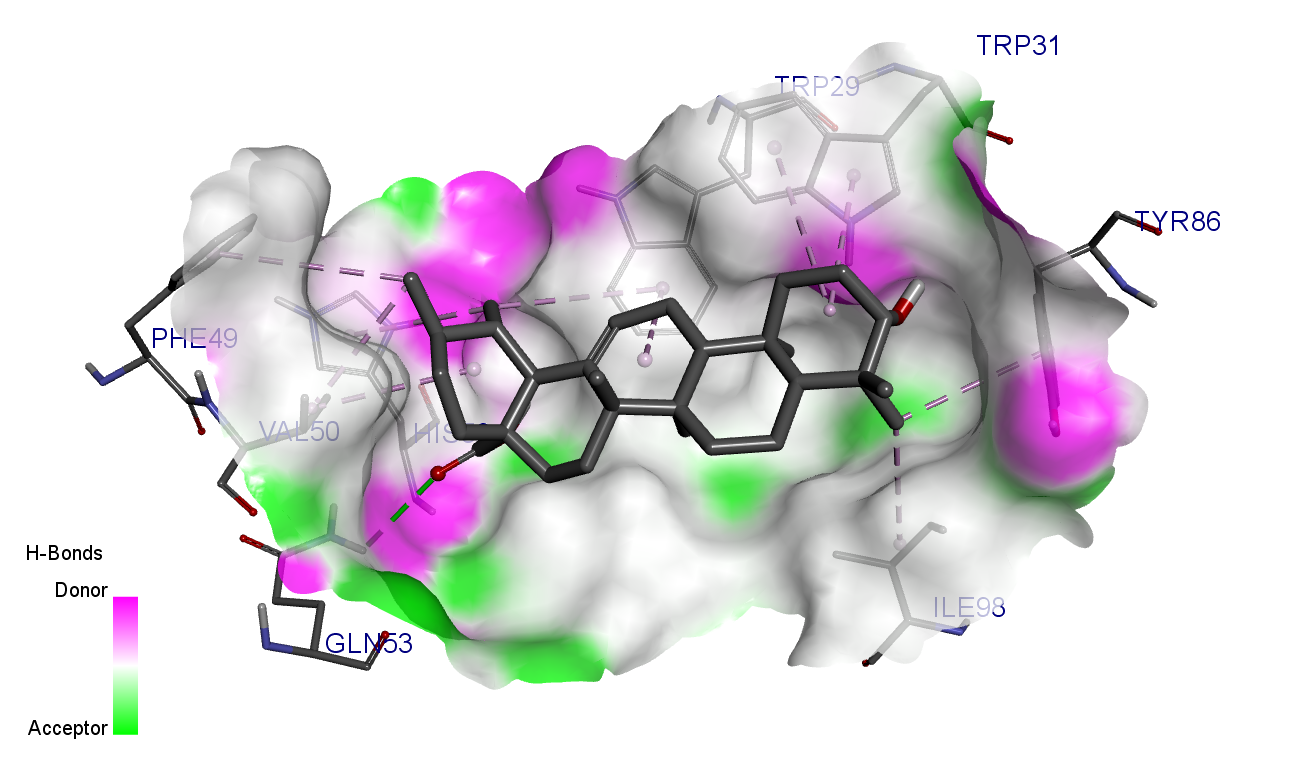
出现新的窗口,点击build H-bond,点击show info,
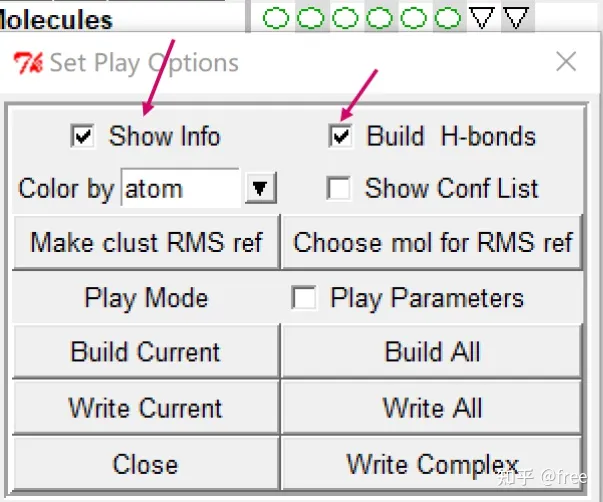
出现新的窗口,得到第一次对接结果的结合能数据,形成氢键个数等等。
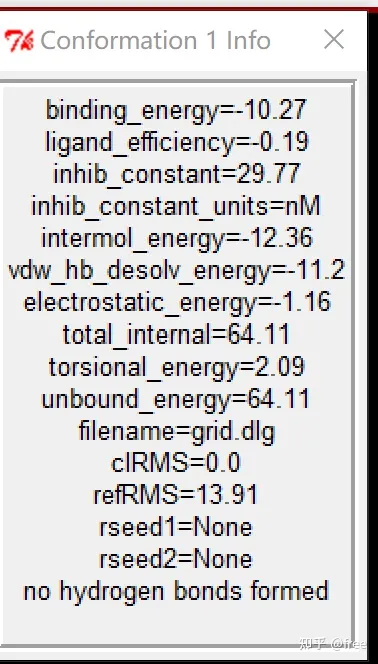
点击analyze-》conformations-》load…查看其它结合能信息
接着,点击write complex,
输出格式为pdbqt的文件,手动输入后缀
保存后,用openbabel将格式转化为pdb格式,接着就可以用pymol(pymol安装看开源版pymol的下载与安装(写给自己) - 知乎 (zhihu.com))打开查看对接结果以及绘图。
*用pymol输出图片*
- 打开pymol,file-》open,选择pdb格式的文件。
2.点击pymol右下角的s,可以显示出氨基酸残基
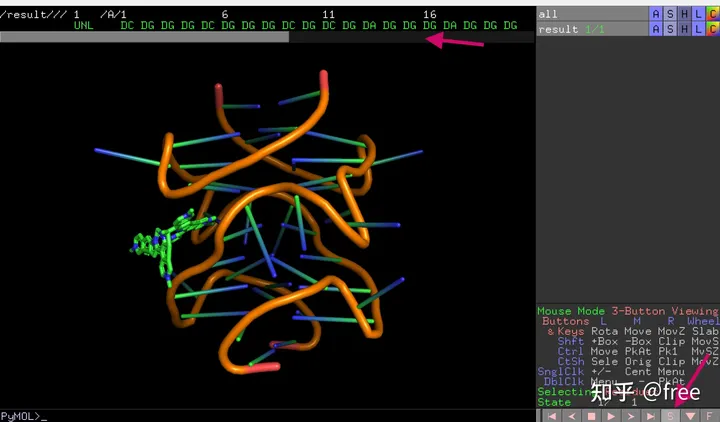
这里的UNL是小分子,后面的是蛋白质残基。